

Introduction
On May 26, 2021, the European Union Medical Device Regulation (EU) 2017/745 (otherwise known as EU-MDR) went into effect for all member states, following a one-year extension due to the COVID-19 pandemic. The implementation of this regulation puts into play transformational requirements for medical device manufacturers, in many cases including enhanced rigour and comprehensiveness for the collection and management of medical device data. The regulation is complex, allowing manufacturers to challenge existing practices, create new processes and introduce a thoroughly managed program that will support a pathway to medical device product compliance.
One key component of MDR compliance is the literature review, a rigorous and systematic approach to searching for evidence-based research that validates medical device product safety and benefit. Literature reviews are performed as comprehensive queries of published data including (but not limited to) scientific papers and studies, resulting in a concentrated data set that supports the product under evaluation by answering a refined research question. For EU-MDR, the literature review supports Clinical Evaluation Reports (CER) and Post-Market Clinical Follow-up (PMCF) reports, as required by Annex XIV, Parts A and B, respectively, in the EU 2017/745 regulation.
Guidelines that strictly outline how a literature summary should be conducted, taking into account compliance with the new EU-MDR, are scarce; however, numerous documents can be referenced when choosing best practices for performing a review, including MEDDEV 2.7/1 Rev 4, which the Medical Device Coordination Group (MDCG) has determined to be the key reference for general guidance on literature review search parameters and protocol. Annex 5 of this document outlines the most critical requirements for the literature review in MDR. In addition, MDCG provides document 2020-13, Clinical Evaluation Assessment Report Template, is primarily aimed at notified bodies and clinical evaluation reviewers. Section D of this document covers the search parameters and literature review.
There are several challenges related to performing a literature review, which will be explored below. Ultimately, all compliant and satisfactory literature reviews employ a combination of efficiency, thoroughness, and repeatability, while leveraging cost-effective solutions for medical device manufacturers. Automated literature review solutions that are designed to address these challenges offer an optimal solution, one that maximizes audit readiness, repeatability and data organization, as well as team resource utilization throughout the process.
An Opportunity for Improving Old Business Practices?
New and Unfamiliar Processes
For many medical device manufacturers, EU-MDR prompts an opportunity to assess and replace outdated and inefficient business processes. For literature reviews, automated and intelligent workflows represent a dramatic change in operations, with smart technology replacing existing partially or fully manual procedures. Increased resource demands have prompted many organizations to hire teams of consultants and staff members, whose jobs are to focus squarely on EU-MDR requirements to ensure compliance. Finding experts who understand them well enough to drive compliant solutions is difficult. Ultimately, new EU-MDR literature review processes represent a significant challenge that most medical device manufacturers must face.
Workload and Resource Constraints
The addition of new regulatory requirements translates into a potential increase in operational costs and the need to hire new resources. It may also significantly impact project timelines. Therefore, the need to make room for the augmented workload and resources introduces substantial challenges to manufacturers by further limiting budgets, timelines and human capital allocation. As a result, driving greater efficiencies by automating literature reviews is critical.
Transparency and Continuous Updates
One key aspect of the literature review process that can pose a challenge is the need to provide auditors with accurate, rigorous records that demonstrate transparency and reproducible results. Maintaining compliance with EU-MDR regulations is an ongoing process that requires manufacturers to consistently monitor for adverse events, to ensure that changes to product safety and risk are appropriately captured and conveyed in advance of the audit process. Many organizations, particularly those that rely on manual literature review solutions, may find this continuous monitoring a challenge, especially in the current era of information/data overload.
Best practices and strategies that address EU-MDR literature review challenges
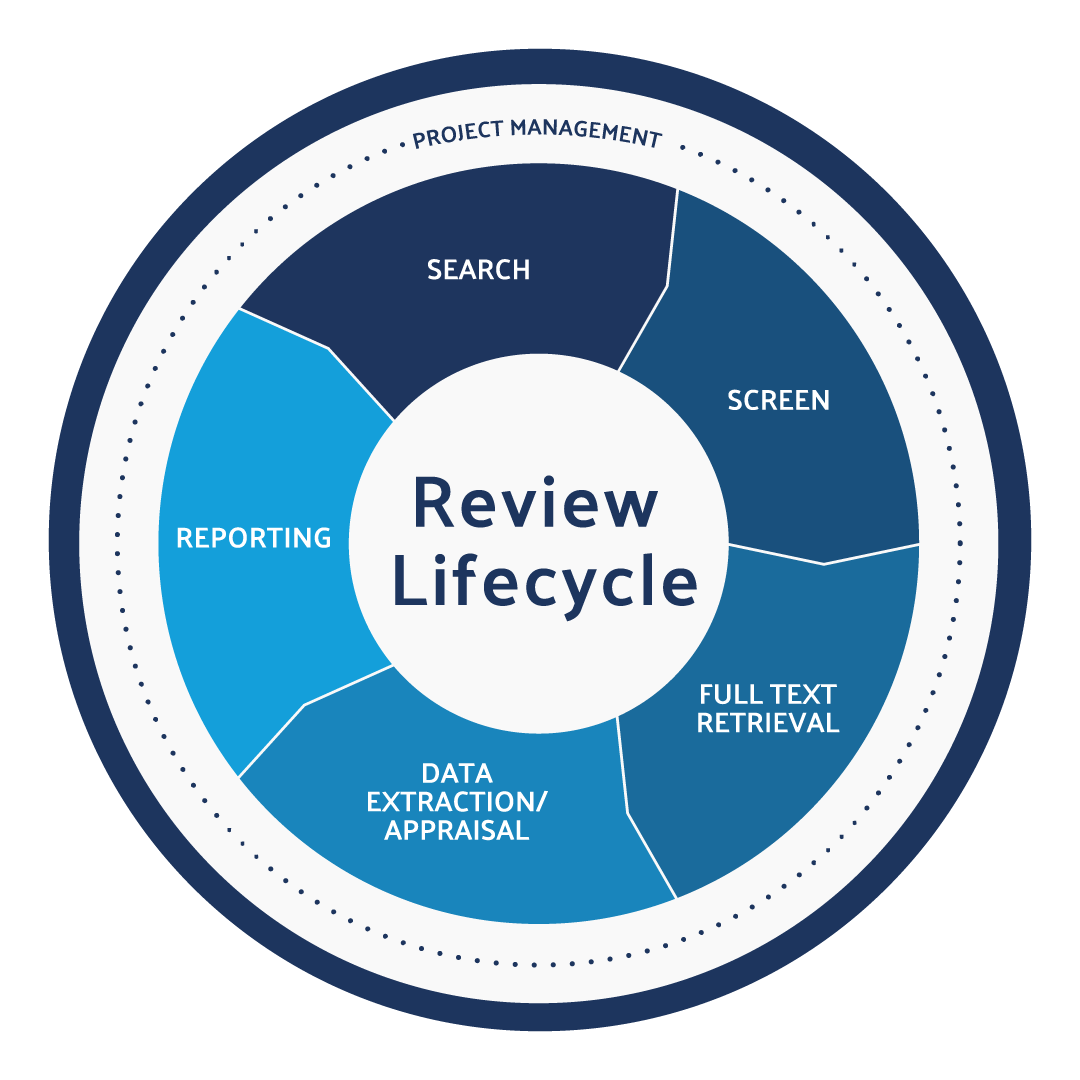
The literature review can be broken down into a lifecycle, with each step employing unique strategies for ensuring compliance:
Define the research question
Defining an appropriately scoped research question is a critical component of the literature review. Research questions that are too broad may lead to a broad set of search criteria and return too many references that will need to be qualified. Too narrow of a research question may fail to retrieve all relevant evidence-based data and capture the full complexity and function of the medical device product. Furthermore, the research question must yield search results that support the requirements of clinical data as laid out in EU 2017/745 Annex XIV, Part A, Sections 1-4. Specifically, clinical data from the literature review process that supports the Clinical Evaluation Report (CER) must include both favorable and unfavorable data and must support the technical, biological, and clinical characteristics of the device.
Defining the research question is the first step in performing a literature summary, but it is also a step that is revisited throughout the literature review lifecycle. MEDDEV 2.7/1 Revision 4, Appendix 5 recommends establishing a literature review protocol that can be used as a key audit tool that will contain the research question. As such, the protocol will define key search terms, databases and sources of data, selection criteria, appraisal and analysis plans and other components that support the Clinical Evaluation and Performance Evaluation Reports. Establishing a literature review protocol using this reference as guidance, and including each of these components combined with a well-defined research question, will set medical device manufacturers up to successfully comply with EU-MDR requirements.
Search relevant databases
Diversifying databases to include multiple source locations and search engines will ensure that all relevant and current clinical data is included in the literature review for the device under evaluation.
For example, healthcare-focused databases like EMBASE or PubMed should be used, in addition to non-EU-based safety databases such as MAUDE (Manufacturer and User Facility Device Experience). The aim here is to identify the most robust and directly relevant data that supports the safety and efficacy of the medical device product.
Furthermore, medical writers must continuously re-run searches across databases for newly published articles over the course of the review. Using a literature review software platform, such as DistillerSR, that can leverage auto-alerts from data providers to keep the growing set of citations automatically up-to-date, eliminates the tedious task of continuously searching for and uploading newly published or updated records.
Screen references for relevance
MEDDEV 2.7/1 Revision 4, Annex 5, Section 3 outlines methods for setting up the literature review screening guideline. This part of the protocol is critical in demonstrating to auditors that manufacturers are following a robust and repeatable process and can justify the inclusion or exclusion of specific data. Literature review platforms can facilitate the automatic tracking of inclusion and exclusion decisions of specific data, and some, such as DistillerSR, use artificial intelligence to double-check excluded references and help prevent erroneous decisions.
Initially, the search will generate numerous results which will need to be triaged or prioritized for relevance to the medical device and research question. As such, screening is a resource-intensive part of the review process, which can be dramatically reduced through the use of literature review applications. For instance, DistillerSR applies AI to duplicate detection and removal, as well as reprioritized unscreened records based on the likelihood of relevance, helping medical writers find relevant references between 40-60% sooner on average than through conventional screening.
Retrieve full-text articles
Reading the full-text entries of all search results would yield an inefficient and time-wasting strategy. Notwithstanding, perusing the abstracts alone for clinical data and clinical information is insufficient to fully characterize the technical, biological and clinical characteristics of a medical device use case. The Medical Device Coordination Group (MDCG) agrees with this notion and proposes that the full-text content of each article is appraised. The methods of full-text retrieval should be outlined in the literature review protocol, which can be performed repeatedly and reviewed quickly during an audit. Software solutions that automatically retrieve full text versions of articles can significantly reduce efforts and time during this stage of the literature review lifecycle.
For example, DistillerSR retrieves all freely available full-text versions of articles automatically, and for those that require purchase, they can easily be procured through direct integrations to Article Galaxy, RightFind, and e-libraries for the lowest possible cost while ensuring repeat purchases do not occur.
Extract and appraise the quality of the data
Qualifying the information retrieved from multiple sources is another crucial step to ensuring that medical device manufacturers fully understand the validity and applicability of evidence-based data. Triaging only the relevant articles and data will streamline processes and improve the literature review quality. As mentioned, MDCG 2020-13, Section D advises that abstracts only be used for first-pass exclusion and recommends full-text reviews during the appraisal phase of the literature review.
Methods for appraising the quality of the data should be registered in the literature review protocol, as outlined in MEDDEV 2.7/1 Revision 4, Annex 5, Section 3. Poor data quality would result in one or more of the following scenarios:
– Lack of information on elementary aspects (disclosure omissions)
– Numbers too small for statistical significance
– Improper statistical methods
– Lack of adequate controls
– Improper collection of mortality and serious adverse events data
– Misinterpretation by the authors, and
– Illegal activities
Standardizing on a comprehensive assessment tool, such as those incorporated in literature review platforms, will ensure all potential data quality shortcomings can clearly be identified and tracked for auditability. DistillerSR comes with a number of industry-standard assessment templates that can be adopted and modified to meet your specific literature review protocol while automatically tracking and associating all decisions and data collected to the appropriate article.
It is important to note that poor data quality does not mean data that shows adverse events. As outlined in EU 2017/745 Annex XIV, Part A, Sections 1-4, the Clinical Evaluation Report (CER) must include both favorable and unfavorable clinical data in order to support the technical, biological and clinical characteristics of the medical device.
Document purpose and methods
The purpose and methods should all be documented in the literature review protocol, as outlined in MEDDEV 2.7/1 Revision 4, Annex 5, Section 3. A robust, easily traceable literature review protocol will ensure repeatability and transparency in audits, increasing the likelihood of compliance. In addition, the protocol can serve as a project management guiding document to drive resources supporting the literature review process.
Literature review platforms help to facilitate adherence to specific protocols, while automatically tracking each action. DistillerSR, for example, makes it easy to view the provenance of every cell of data and ensures complete transparency and auditability of the entire process, which enables a fully defensible, transparent and repeatable review.
Monitor for new content and adjust the research question if necessary.
The research question supports product safety and clinical use but may need to be revised in the event of new adverse events or literature content. As a result, guidelines listed in MEDDEV 2.7/1 Revision 4, Annex 5 are best followed to ensure the search provides clinical data on current interventions. The research question may need to be adjusted, thereby revisiting the literature review lifecycle. Here lies the opportunity for employing automation tools that enable reutilization of previously-stored search criteria and build a reference library in order to avoid unnecessary repetition and wasted resources and time.
Conclusion
The complexity, resource-heavy and time-consuming nature of the literature review requirements as set in the regulations listed above cannot be overlooked. Leveraging tools such as automation software solutions, like DistillerSR, that allow for the most efficient resource allocation can serve as a valuable strategy to significantly streamline the time to compliance. When in doubt, referencing the relevant sections of MEDDEV 2.7/1 Revision 4 and adhering to the guidelines and examples established can help drive best practices, ensuring that medical device manufacturers increase their likelihood of complying with the rigorous criteria established by EU-MDR.




