

* This post has been updated on April 30, 2020 to reflect the extension of the EU MDR deadline due to the COVID-19 pandemic.
In April 2017, the European Parliament and Council adopted two new Medical Device Regulations (MDRs). Regulation 2017/745 sets quality and safety standards for devices other than in vitro diagnostics, which are now covered by MDR 2017/746 (commonly referred to as IVDR).
Implementation of these directives is fast approaching, and there will be few device manufacturers not impacted by the new legislation.
As these new MDRs provide a significant overhaul over prior legislation, medical device manufacturers need to be armed with the necessary information to ensure they are compliant. This quick guide explains the aims of the legislation, highlights the key changes from previous regulations, and flags some important considerations as you prepare.
What are the aims of the new medical device regulations?
One of the key aims of the new legislation is to improve patient safety by modernizing and tightening EU medical device approval and post-marketing requirements.
The MDRs are also intended to consolidate and strengthen previous regulations and allow for more concise and sustainable interpretation, as well as transparency and traceability between manufacturers and Notified Bodies (the device conformity assessment reviewers).
Ultimately, both directives are designed to harmonize the regulation of medical devices across EU Member States and facilitate trade.
What are the main changes in the new regulations?
In terms of timelines, the new regulations were passed in 2017, but provided for a three-year transition period for medical devices. The deadline for MDR implementation is May 26, 2021, with an additional two years allowed for IVDR.
There are several changes that these regulations will bring in, including:
Modification of the medical device definition to include devices not previously falling under the definition.
Classification of in-vitro diagnostics into four distinct classes, requiring Notified Body review for most.
Increased vigilance and market surveillance, including collection of performance data post-marketing and assignment of a unique device identifier (UDI).
Facilitation of the Eudamed database for pre- and post-approval product information, including clinical evaluation conclusions.
Tighter pre-market controls on high-risk devices, requiring clinical evaluation and clinical investigation.
Creation of a risk-based classification system, including in-vitro diagnostics and products previously exempt from the regulations.
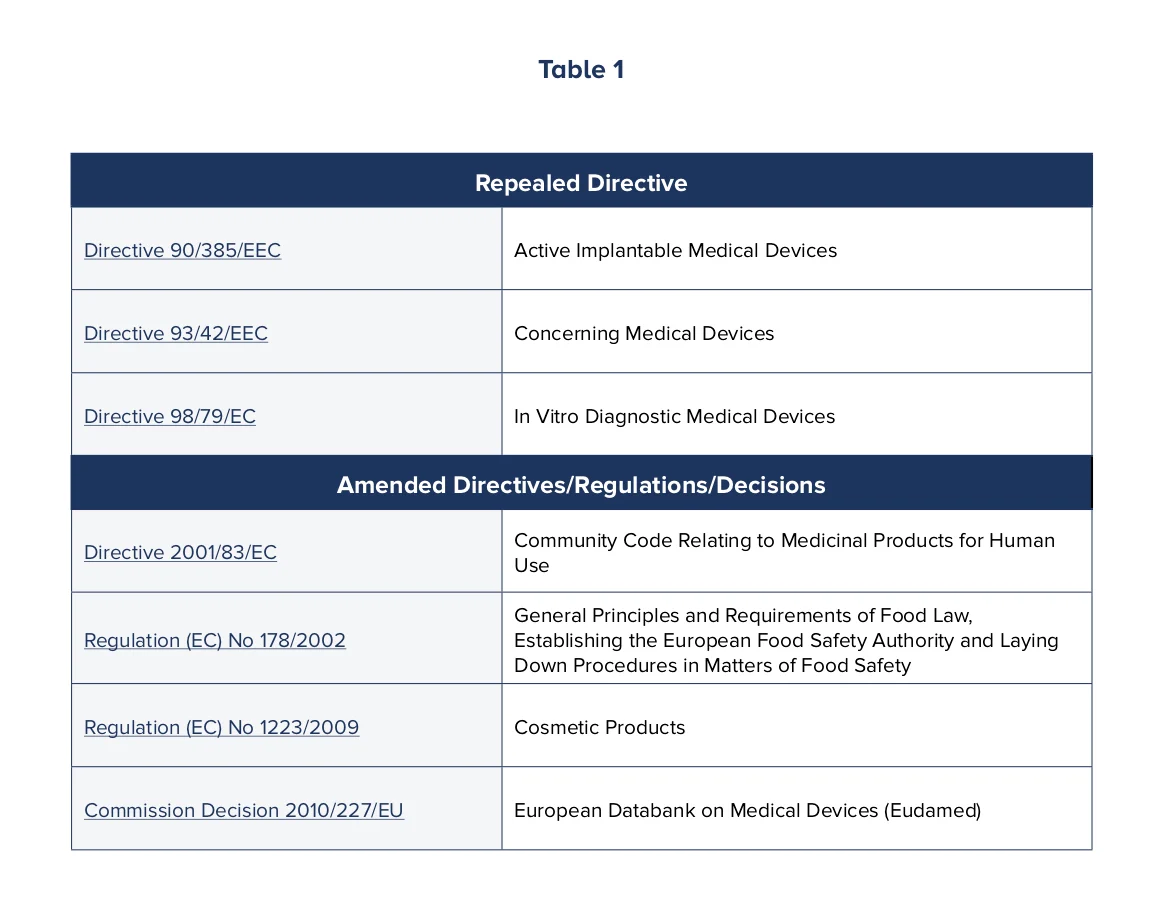
One of the issues in the past was that regulation of medical devices fell under multiple directives, regulations and decisions, which caused confusion. These definitions have now been modernized to accommodate innovative device technology and uses, with additional focus placed on the implementation and use of the European device databank, Eudamed, by Member States, and in some cases, healthcare professionals and the public.
Table 1 lists the directives, regulations and decisions that have been amended or repealed as a result of the new MDRs. (Please click on the table to view more details.)
What do device manufacturers need to do to prepare?
The new MDRs affect manufacturers of new medical devices as well as those with devices in the European market (those already CE marked) prior to May 26, 2020. While the regulations are not intended to be retroactive, existing devices will need to re-certified according to the new MDRs when applying for new intended uses.
As with any new legislation, there is a laundry list of requirements for device manufacturers, which includes:
- Establishing a Post Marketing Surveillance System (PMSS) in line with the clinical evaluation/level of device risk.
- Assigning a Responsible Person.
- Ensuring there are sufficient financial compensation measures in place to cover potential liability.
Manufacturers should expect implementing MDR requirements to be complex and expensive. With more devices being escalated on the risk scale, a higher level of effort will need to be made to prove safety and efficacy, including, for some devices, the conduct of clinical trials.
Due to a surge in demand, medical writers and regulatory experts with clinical evaluation report (CER) experience are becoming increasingly difficult to hire. Implementing software-based systems that can increase the capacity, throughput and maintainability of CER processes, without a corresponding increase in headcount, has become an imperative.
With the deadline now less than a year away, choosing, implementing and training on new software systems that can automate components of the literature review process should already be underway. Decisions on whether to build such systems internally, or license existing systems, will impact both budget and timelines.
Technical documentation, clinical data and labeling will all need to be updated. Many devices will need more robust documentation than previously required for conformity assessment, such as clinical investigation plans (CIPs) and clinical evaluation reports (CERs). Additionally, all devices will need to be assigned a UDI.
Manufacturers will also need to accommodate internal changes, which may include hiring additional staff including a Responsible Person, building or licensing compliant vigilance, tracking and licensing software for safety reporting and managing clinical evaluation reports, and writing processes (for example, standard operating procedures).
What guidance or help is available?
No doubt, there is a lot to think about and do when it comes to preparing for the new MDRs, but luckily there are some easy steps you can take to get prepared.
First and foremost, make sure you’ve reviewed the guidance and information documents posted by The European Commission. These are designed to assist manufacturers in interpreting the regulations.
Still overwhelmed? Consider enlisting the help of experts in specific aspects of the implementation, especially where your existing processes are not designed to meet the new expectations. You may also want to look at adopting new processes and technology solutions to address the increased data management challenges as a result of the new MDRs.
The countdown to May 2021 is on. As the industry works to understand and adapt to the new reality, it’s becoming increasingly apparent that the status quo will need to be adjusted to meet the requirements.




