

In a recent webinar, DistillerSR customers Monique Liston, Senior Medical Writer at NuVasive and Shelley Jambresic, Senior Clinical Evaluations Manager at Geistlich Pharma were joined by Dr. Bassil Akra, founder and CEO of Akra Team GmbH, Dr. Julien Senac, Global Director IVDR at TÜV SÜD, and David Kovac, DistillerSR Account Executive to discuss the challenges of developing a methodologically sound state of the art framework while keeping up with the latest literature and tight regulatory deadlines.
Q: It seems state of the art has been in discussion since EU MDR was introduced, back in 2017. Dr. Bassil Akra, why is it such an important topic for medical device manufacturers?
A: When people hear state of the art, they recognize it is not a straightforward definition, the term itself can be interpreted in many ways. Generally speaking, state of the art refers to cutting edge technology. In the context of medical devices, however, EU MDR defines state of the art as the benchmark/current state of what is currently available in the market, compared to your medical device. The objective of the state of the art is to demonstrate the acceptability of a new medical device being introduced in the market, for example.
Q: Let’s turn to Dr. Julien Senac for the notified body perspective. What are some of the challenges notified bodies are experiencing at the moment?
A: The biggest ongoing challenge is capacity related to recruiting qualified staff. Finding people with the right level of expertise is difficult. Furthermore, the staff initially was going through a steep learning curve, dealing with requests for information from medical device manufacturers while trying to understand the evolving set of rules outlined in the MDR. As a result, our ability to process applications in that scenario was limited. We have been making strides in terms of capacity but it remains a challenge. The message we have for medical device manufacturers is to get ready early so notified bodies can allocate their own resources accordingly.
Q: How do notified bodies define state of the art? What is your expectation?
A: One of the topics I have been discussing frequently refers to the intended purpose definition. State of the art is defined by the intended purpose of the medical device, it is based on how your product is used and in which conditions. I will take an IVD example to illustrate: let’s say tomorrow I come up with a new COVID-19 15-min rapid test. I want to introduce a new technology. What should I set as the benchmark for my device that is currently being used? Do I compare my new test to the laboratory PCR test, which is considered the best way to determine whether a patient has COVID or not, or do I compare my new test to the parking lot, rapid test? As we know, the laboratory test will take one or two days. The rapid test is designed for quick turnaround. That’s where you would look at your technology and intended purpose combined with the intended setting. Your new test is a 15-minute test, it is designed to give a quick answer and to be administered in an informal point of care, like a parking lot or walk-in clinic. So your state of the art benchmark is the rapid test. State of the art does not mean cutting edge technology, it means the widely accepted technology in the market. Maybe your new device will set a new technology parameter once it is in the market. The notified body expects you to demonstrate with robust evidence that your new medical device works within the parameters of medical devices that have received the regulatory seal of approval and match your intended purpose and setting.

Q: Shelley, you work with clinical evaluations at Geistlich Pharma. How are you dealing with the challenges and opportunities from establishing state of the art for your medical devices?
A: The state of the art is basically the picture frame where you place your device. It is not just a necessary evil but it will also provide you with valuable guidance early in the medical device lifecycle. On one hand if you have a legacy device, state of the art will provide you with the acceptance criteria to present your device within the correct parameters. On the other hand if you are about to develop a new device, state of the art will guide your approach, for example, to collect your clinical data. When you establish your state of the art benchmark, you get safety and performance-related clinical parameters. And those will basically correspond to the outcomes in your clinical investigation. The most important aspect though is not to lose focus from what is relevant for the patient, especially when it relates to the performance of your device. Make sure you compare outcomes that were measured in a clinical research setting with outcomes measured in the field to determine what is meaningful to your performance parameters. For example, in a dental implant case, when you look at the state of the art benchmark today and how the outcomes are measured, there is not a lot of data related to the performance of the device. There’s significant data related to bone density but this particular outcome is irrelevant for the patient. The patient cares about whether the dental implant is stable, healthy and will last.
Q: So from what you’re telling us, state of the art will give us guidance to establish the key indicators for safety and performance. Monique, what role does state of the art have at NuVasive?
A: From our perspective, state of the art establishes the role of your device within the clinical landscape, compared to equivalent devices. First, we identify competitive devices and practice guidelines for alternative therapies currently available to surgeons and patients in the context of the safety and performance of our device. From there we get to the benchmark and acceptance criteria for how our device should be performing. The way you think about state of the art matters. It is important to fully appreciate and understand the role of state of the art in clinical evaluations. As Shelley mentioned, it is not designed to be a checkmark in the regulatory submission process, it is a continuous process to ensure medical devices are complying with rigorous industry standards. It is an opportunity to step back and look at the complete body of evidence from currently published literature, assess the standard of care, the options available to surgeons and patients and compare it with your device. The safety and performance outcomes obtained from your established state of the art benchmark will allow you to continuously demonstrate that your device is safe and effective compared to standard of care and competitive devices.
Q: State of the art is a critical, continuous process, not a single shot at hitting the regulatory mark. And it is not based on subjective criteria, it is based on evidence collected from published literature and real-world data. Shelley, can you elaborate on the challenges related to continuous compliance?
A: The robust justification requirement introduced by EU MDR translates into the need for gathering enough evidence to support your state of the art safety & performance claims so you have to go beyond your routine benchmarks. So the biggest challenge to achieve continuous compliance is keeping up with scientific literature. Furthermore, you‘re trying to strike a balance between cherry picking and casting a wide net with your initial literature review. You need to set your search terms carefully so that you end up with sufficient relevant results. Finding the right keywords and avoiding bias are critical at the start of the evidence gathering process.
Q: Monique, what is your experience?
A: Adding to what Shelley explained, your initial search is the foundation for state of the art. It could be easily brushed off but for our team at NuVasive it is the crux of the entire clinical evaluation process. It is important for medical writers to understand the databases they are searching from. Each database works with different syntax, different field tags, and different codes. These are the things that can either make or break your search because they affect your precision. We don’t have time to screen thousands of irrelevant articles. I agree with Shelley that your search keywords are also really important. You don’t want to bias your search so you need to be broad and specific at the same time to ensure you’re capturing all of the alternative treatments that are relevant for your intended purpose. My suggestion is to iterate your search terms to match your intended purpose and set time aside to meet with surgeons and doctors if they are available resources.
Q: When I worked at a notified body, I always advised people to tell a logical, digestible story. What is your advice, Dr. Senac?
A: My advice is to establish your state of the art benchmark as the starting point for your medical device. Look at what’s out there in the market, think about what you are trying to achieve with your device and integrate your state of the art safety and performance outcomes into your design process. When manufacturers delay this state of the art comparison, they end up trying to match their intended purpose to the competitive benchmark and introduce inconsistencies into their reporting processes.
Q: Why do notified bodies raise so many questions about state of the art?
A: It all depends on the data that is provided. When we look at the state of the art, we are looking for a balanced approach. We will analyze your submitted methodology and rerun your search to see if we are able to get the same results. We will then look at the outcome you have set in your report and whether the weight of the studies you included as supporting evidence is enough and accurate. We need to be sure that we have an exact and most accurate picture of the state of the art at that moment. In some cases, when we challenge your report, it may be because your answer was not clear or because your answer was not complete. Either way, it doesn’t mean you have to redo everything in these cases. It goes back to telling a clear story and ensuring you have enough data to back up your conclusion. Sometimes you think you told a good story but it is not actually well received by the notified body.
Q: In terms of best practices, Shelley, what would you recommend to reduce the number of questions related to state of the art submissions?
A: There is no such thing as a perfect submission so the focus needs to be on avoiding questions from the notified body. The simplest way to achieve this is to make sure your story makes sense. Sometimes you’re so deep into the topic and working under such time pressure that you tend to miss important details. Make sure you have quality control built into your process so you don’t overlook the critical data. The other piece of advice I would give is to look at the guidance available. Use the documentation provided by MCDG and the checklists provided by notified bodies. Get familiarized with the language and the formulations they expect to see in your submission.
Q: Shelley, why is literature key to establishing state of the art?
A: The literature is key for you to demonstrate the intended purpose for your medical device. It is what will support your performance and safety claims to the notified body. There are vast volumes of published literature out there and the only way to narrow it down to your intended purpose is to conduct a systematic search that is structured and reproducible. It is important to note that if your device has a novel/innovative purpose, you will not find literature to justify your intended purpose and consequently, you will not have a benchmark to establish state of the art. In this case, it will come down to a meaningful justification to introduce such a device in the market backed by a robust risk-benefit analysis.
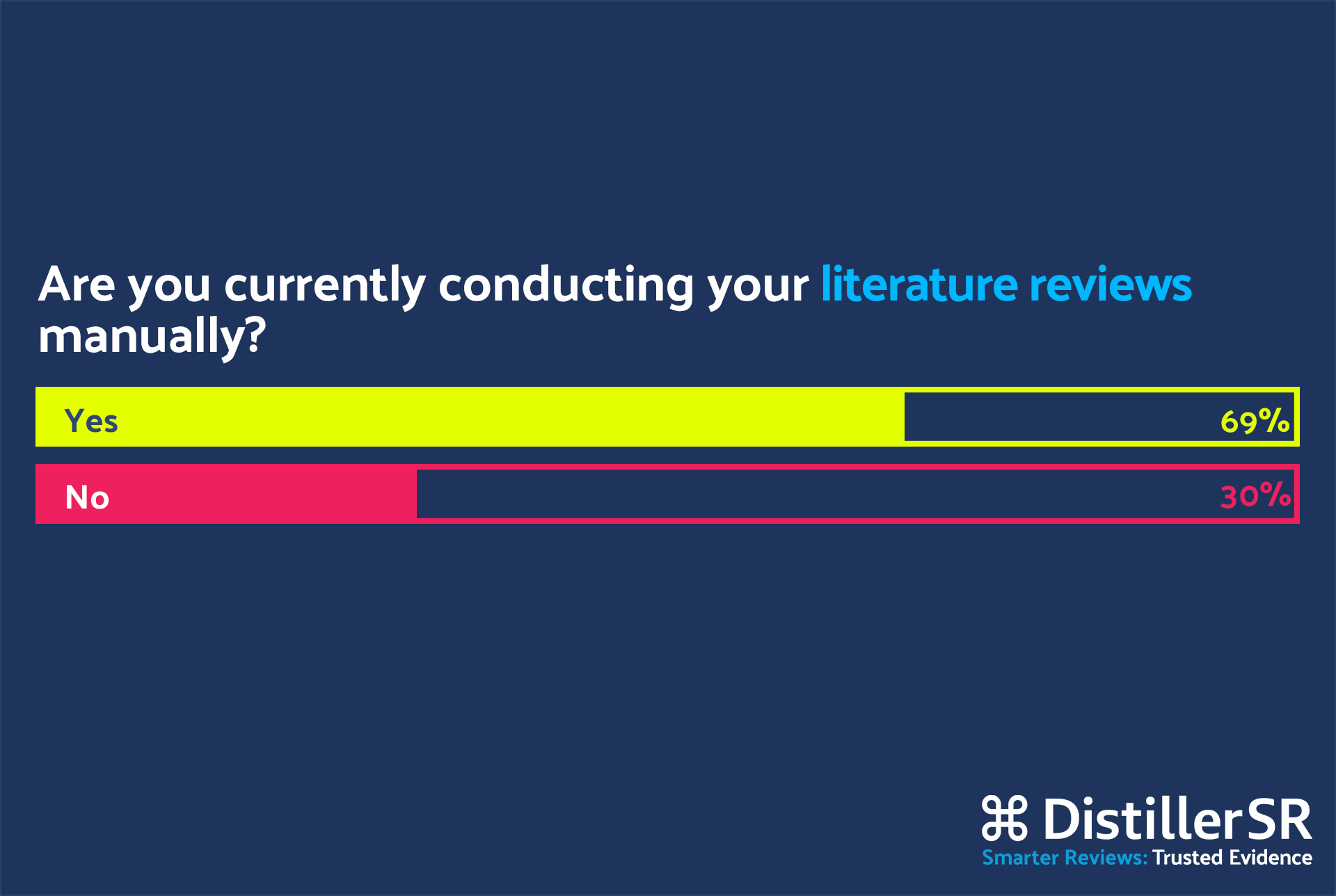
Q: Monique, looking at the poll results, 70% of the attendees are still conducting their literature reviews manually while only 30% of them are employing automation. Can you tell us a bit about your experience? What are you currently using to improve your efficiency while conducting literature reviews?
A: There are many solutions available in the market so finding a platform that fits your team and your project requirements is critical to realize much needed efficiencies in the literature review process. Learning a new platform is time consuming so you also need to have a team who is willing to test and trial these different softwares until you find the best fit. We are using DistillerSR because it fits all the boxes for us. It has become the single source of truth for our entire team and medical device portfolio. It allows the team to work from a consistent study assessment criterion across all of our projects. In the end our outputs look very similar. For the search results, we have built custom forms inside DistillerSR to ensure we are capturing all the data elements that will be addressed in the clinical evaluation process. These forms can be easily exported to Excel and they correspond to the complete evidence behind our clinical evaluations. So our main goal is to streamline the literature review process, saving time and ensuring consistency. Another important goal is to modularize our data, which means we are reusing previously collected datasets as much as possible to relieve the burden of work duplication. For example, we look at the work done for device A and see if the indication applies to device B consequently saving significant time not digging into the same data sets and unnecessarily repeating the screening and data extraction processes. We quickly realized that data reuse was something that would benefit the entire organization, not only the medical writers and clinical evaluation team.
“We are using DistillerSR because it fits all the boxes for us. It has become the single source of truth for our entire team and medical device portfolio. It allows the team to work from a consistent study assessment criterion across all of our projects. In the end our outputs look very similar. For the search results, we have built custom forms inside DistillerSR to ensure we are capturing all the data elements that will be addressed in the clinical evaluation process. These forms can be easily exported to Excel and they correspond to the complete evidence behind our clinical evaluations.”
Q: David, could you briefly explain the concept of data reuse for manufacturers establishing state of the art?
A: State of the art is a living review that you have to keep constantly updated to ensure you’re looking at the most up to date literature reflecting what is available in the market in terms of equivalent therapies. The main benefit of data reuse in this context is the ability to quickly identify what you have already reviewed, screened, and appraised in the past as not to duplicate efforts. For a manufacturer with a wide portfolio, the extended benefit is to reuse the same evidence dataset for similar medical devices, as mentioned by Monique and Shelley. Essentially, when you run a state of the art literature review for the first time using a literature review management software, you are building an evidence database that can be reused by multiple team members for devices with a similar intended purpose. This database will be continuously updated ensuring you and your teams are keeping up with the latest evidence, saving time and effort in the compliance process. DistillerSR has a module called CuratorCR that centrally and dynamically manages an organization’s evidence-based research, allowing you to continuously curate, share, update and reuse its data.
“Essentially, when you run a state of the art literature review for the first time using a literature review management software, you are building an evidence database that can be reused by multiple team members for devices with a similar intended purpose. This database will be continuously updated ensuring you and your teams are keeping up with the latest evidence, saving time and effort in the compliance process.”
Q: What roadblocks did you encounter while trying to implement a literature review automation platform in your organization, Shelley?
A: Making sure you have a team who is willing to experiment with new tools and embrace change is definitely important, as Monique mentioned, but to me the main issue was to convince my management team to pay for new software. The decision is always about time and money. So I sat down with a timer and actually timed every step of my literature review, manually versus with DistillerSR. In the first three months of the proof of concept phase, we completed the literature reviews 40% faster with DistillerSR. It is even faster now that the platform is fully implemented. For title and abstract screening, for example, it takes me 5 to 10 seconds to include or exclude a reference by using the DistillerSR keyword highlighting functionality. Being able to audit the records related to any medical device was also critical to my management team when they were making a decision on whether DistillerSR was the right platform for our team. If I am gone, the next person will be able to pick up the work where I left off. Furthermore, the ability to share data across departments, for example, our marketing, sales and clinical operation teams can easily extract data such as end points for their own purposes.
“I sat down with a timer and actually timed every step of my literature review, manually versus with DistillerSR. In the first three months of the proof of concept phase, we completed the literature reviews 40% faster with DistillerSR. It is even faster now that the platform is fully implemented. For title and abstract screening, for example, it takes me 5 to 10 seconds to include or exclude a reference by using the DistillerSR keyword highlighting functionality.”




